What is Apert Syndrome?

Apert Syndrome (AS), also known as acrocephalosyndactyly, is a rare genetic disease characterized by skeletal abnormalities that begin during prenatal development [1]. AS is caused by a mutation in the Fibroblast Growth Factor Receptor 2 gene (FGFR2) which is involved in many important processes such as cell growth and division (proliferation), cell maturation (differentiation), and bone development [2]. The mutation leads to increased cell signaling causing cells to mature too quickly which promotes the premature fusion of bones [3].
Life with Apert Syndrome
|
|
|
What are the symptoms?
Apert Syndrome is characterized by craniosynostosis, a condition that involves the premature fusion of one or more cranial sutures, and syndactyly, the fusion of fingers and toes [1]. Additionally, many individuals with AS display a tall or peaked head shape (acrocephaly) as well as some key facial features including a sunken appearance in the middle of the face (midface hypoplasia), wide-set eyes (hypertelorism), an underdeveloped jaw, and a "beaked" nose [3]. Secondary complications often associated with AS include, elevated pressure in the brain, damage to corneas, hearing loss, dental abnormalities, obstructive sleep apnea, cleft palate, varying degrees of developmental delay, and mild to moderate intellectual disability [5].


|


|


|


|
How is it diagnosed?
Since Apert Syndrome develops during embryonic growth, individuals with AS are most commonly diagnosed at birth or during infancy. To formally make a diagnosis, a doctor will look for the characteristic bone abnormalities affecting the head, face, hands, and feet. They may also perform a skull radiograph, CT scan, or MRI of the head to determine the nature of the bone abnormalities [4]. Individuals may also have testing for mutations in the FGFR2 gene, which can provide a genetic diagnosis. In some instances, features of AS may be detected before birth. This would be done through prenatal 2D or 3D ultrasound or fetal MRI [3].
How is it Inherited?
|
Apert Syndrome is a rare disease that results from a randomly occurring, sporadic mutation. Nearly all cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or in early embryonic development. These cases occur in people with no family history of the disorder [2]. Although the mutation occurs for unknown reasons, studied have found an association between sporadic cases and increased paternal age [6]. AS is estimated to occur in about one in 65,000 births. Males and females seem to have the condition in relatively equal numbers [3]. Rarely are there cases in which AS is passed on within a family; however, in those situations it is inherited in an autosomal dominant fashion. Dominant genetic disorders occur when only a single copy of a mutation is necessary to cause a particular disease. The risk of passing the mutation from an affected parent to an offspring is 50% for each pregnancy.
|

Fig. 1: Apert Syndrome most commonly results from de novo mutations. In rare cases when the disease is passed on within a family, it is inherited in an autosomal dominant fashion.
Created in BioRender.com
|
What is the role of the FGFR2 gene and protein?
|
Apert Syndrome is caused by a genetic mutation in the FGFR2 gene. This gene is located on chromosome 10 on the long arm at 10q26.13,and encodes a fibroblast growth factor receptor 2 protein that is involved in important processes such as cell growth and division (proliferation), cell maturation (differentiation), bone development, formation of blood vessels (angiogenesis), wound healing, and embryonic development [2]. There are two point mutations that change one of the amino acids in the protein sequences and lead to AS. The mutations are designated as “Ser252Trp” and “Pro253Arg" [3]. The first mutation is the most common and is associated with severe craniofacial anomalies while the other mutation contributes to severe syndactyly [1].
FGFR2 is a transmembrane receptor protein that interacts with specific growth factors outside of the cell. When growth factors bind to the receptor, it triggers a series of chemical reactions inside the cell that instruct the cell to undergo certain changes, such as maturing to take on specialized functions. For example, this protein signals certain immature cells in the developing embryo to become bone cells and form the head, hands, feet, and other tissues [2]. In the case of the mutated form of the protein responsible for AS, FGFR2 has an increased affinity and altered specificity of fibroblast growth factor (FGF) ligand binding which leads to increased cell signaling and ultimately premature fusion of the bones [1]. |

Fig. 2: FGFR2 gene is located on chromosome 10 at 10q26.13 and has 21 exons.
|

Fig. 3: FGFR2 is a transmembrane receptor protein that binds the fibroblast growth factor ligands. Upon binding, it causes a cell signaling cascade that causes immature cells to differentiate [8].
|

Fig. 4: Mutations in the FGFR2 gene lead to altered ligand affinity around the third immunoglobulin-like domain and altered kinase activity [8].
|
What treatments are available?
There is no known cure for Apert Syndrome. The symptoms and severity vary by individual which leads to a unique treatment plan for every AS diagnosis. Management of the condition is a team-based approach requiring many specialists including pediatricians, neurosurgeons, plastic surgeons, craniofacial surgeons, ophthalmologists, dentists, and others [7]. The current standard of treatment is through corrective surgery. Surgery is required to prevent complete coronal suture closure and protect brain development. These surgeries affecting the brain, skull and face typically are done in stages, but the timing of surgeries as well as techniques used depend upon each child's symptoms, growth, and psychosocial development [3]. Similarly, there is no standard of care for the treatment of syndactyly, fusion of fingers and toes, but multiple surgical revisions are likely as the child grows [7].
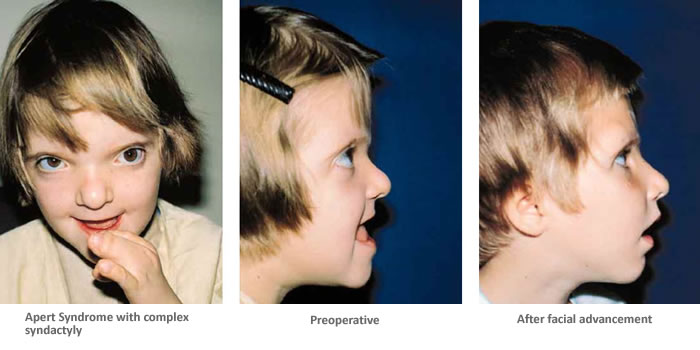
|
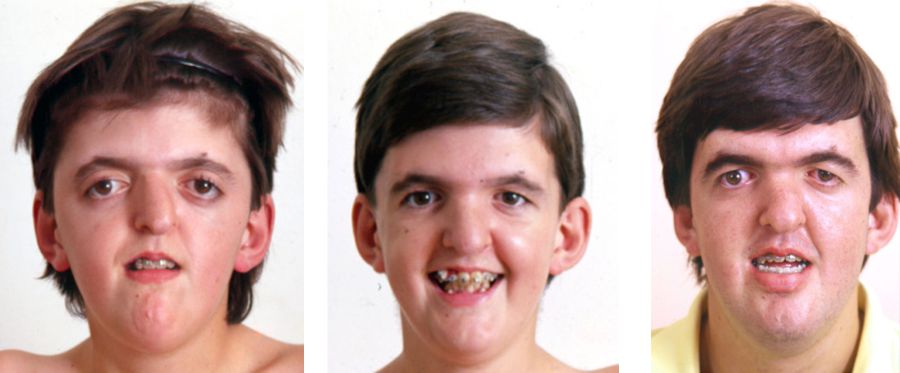
|
|
Common Surgeries:
Crainosyntosis release also called cranial vault expansion is a surgery to treat the fusion of the skull bones too early in development. This typically occurs at 6 to 12 months of age if the infant has normal pressure inside the skull. It can be done earlier if there is increased pressure. In some cases, the surgeon performs an initial procedure in the first year of life to increase space within the skull before the release, in which case the release may be done after 12 months of age [3]. Midface advancement corrects the skull or face for problems with a sunken appearance of the face, abnormal positioning of the eyes, and for abnormal shape of the skull. The timing of this surgery varies. For some children, this procedure is recommended as early as 4 years of age whereas other do not get this surgery until late adolescence. Facial growth does play a role in the longterm success of the surgery and often times multiple surgeries are needed over time [3]. Hypertelorism correction reduces the distance between widely-spaced eyes. This can be done by removing part of the bone between the eyes (interorbital bone), and repositioning the eye sockets closer together for improved appearance and better vision [3]. Separation of fingers and toes is typically done around 9-12 months of age and requires multiple surgeries [3]. |




|
What is the gap in knowledge?
Apert Syndrome is still not well understood. Many of the molecular features exhibited in this syndrome are still unexplainable and require intensive research. Due to the genetic heterogeneity seen in this condition, suspected cases require to be thoroughly investigated by both clinical and molecular methods. Many of the clinical features observed are still unexplainable and thus a definitive treatment plan that can improve the quality of patient’s life still does not exist [1]. What I would like to focus more on through my research project is to better understand the role of FGFR2 in cell proliferation in bone formation. I will be using genomic and proteomic approaches to investigate its this process.
Organizations

|
|

|
References:
[1] Das, S., & Munshi, A. (2018). Research advances in Apert syndrome. Journal of oral biology and craniofacial research, 8(3), 194–199. https://doi.org/10.1016/j.jobcr.2017.05.006 Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6107905/
[2] FGFR2 gene: MedlinePlus Genetics. (2020, August 18). Retrieved from: https://medlineplus.gov/genetics/gene/fgfr2/
[3] Apert syndrome. (2017, April 10). Retrieved from: https://rarediseases.info.nih.gov/diseases/5833/apert-syndrome
[4]Apert syndrome: SYMPTOMS, treatment, and prognosis. (2018, February 13). Retrieved from: https://www.medicalnewstoday.com/articles/320907#overview
[5] Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. 1998 Oct 20 [Updated 2020 Apr 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Retrieved from: https://www.ncbi.nlm.nih.gov/books/NBK1455/
[6] Goriely, A., & Wilkie, A. O. (2012). Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. American journal of human genetics, 90(2), 175–200. https://doi.org/10.1016/j.ajhg.2011.12.017 Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3276674/
[7] Conrady CD, Patel BC, Sharma S. Apert Syndrome. [Updated 2020 Aug 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Retrieved from: https://www.ncbi.nlm.nih.gov/books/NBK518993/
[8]Katoh, M. FGFR2 Abnormalities Underlie a Spectrum of Bone, Skin, and Cancer Pathologies. Journal of Investigative Dermatology,
Volume 129, Issue 8, 2009, Pg. 1861-1867, ISSN 0022-202X, Retrieved from: https://www.sciencedirect.com/science/article/pii/S0022202X15344328www.sciencedirect.com/science/article/pii/S0022202X15344328
Header image: Campeau P, Schlesinger AE. Skeletal Dysplasias. [Updated 2017 Jan 30]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279130/
[1] Das, S., & Munshi, A. (2018). Research advances in Apert syndrome. Journal of oral biology and craniofacial research, 8(3), 194–199. https://doi.org/10.1016/j.jobcr.2017.05.006 Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6107905/
[2] FGFR2 gene: MedlinePlus Genetics. (2020, August 18). Retrieved from: https://medlineplus.gov/genetics/gene/fgfr2/
[3] Apert syndrome. (2017, April 10). Retrieved from: https://rarediseases.info.nih.gov/diseases/5833/apert-syndrome
[4]Apert syndrome: SYMPTOMS, treatment, and prognosis. (2018, February 13). Retrieved from: https://www.medicalnewstoday.com/articles/320907#overview
[5] Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. 1998 Oct 20 [Updated 2020 Apr 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Retrieved from: https://www.ncbi.nlm.nih.gov/books/NBK1455/
[6] Goriely, A., & Wilkie, A. O. (2012). Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. American journal of human genetics, 90(2), 175–200. https://doi.org/10.1016/j.ajhg.2011.12.017 Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3276674/
[7] Conrady CD, Patel BC, Sharma S. Apert Syndrome. [Updated 2020 Aug 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Retrieved from: https://www.ncbi.nlm.nih.gov/books/NBK518993/
[8]Katoh, M. FGFR2 Abnormalities Underlie a Spectrum of Bone, Skin, and Cancer Pathologies. Journal of Investigative Dermatology,
Volume 129, Issue 8, 2009, Pg. 1861-1867, ISSN 0022-202X, Retrieved from: https://www.sciencedirect.com/science/article/pii/S0022202X15344328www.sciencedirect.com/science/article/pii/S0022202X15344328
Header image: Campeau P, Schlesinger AE. Skeletal Dysplasias. [Updated 2017 Jan 30]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279130/
|
Sophia Lenzmeier University of Wisconsin-Madison [email protected] [email protected] Last updated: 5/6/2021 |
|
